What We Do
Research Directions
We build principled computational frameworks — spanning Bayesian statistics, deep learning, and modern AI — that bridge statistical theory and biological discovery, operating across scales from molecules to tissues.
⚗️
Bayesian Transcriptome Deconvolution and Gene Regulation
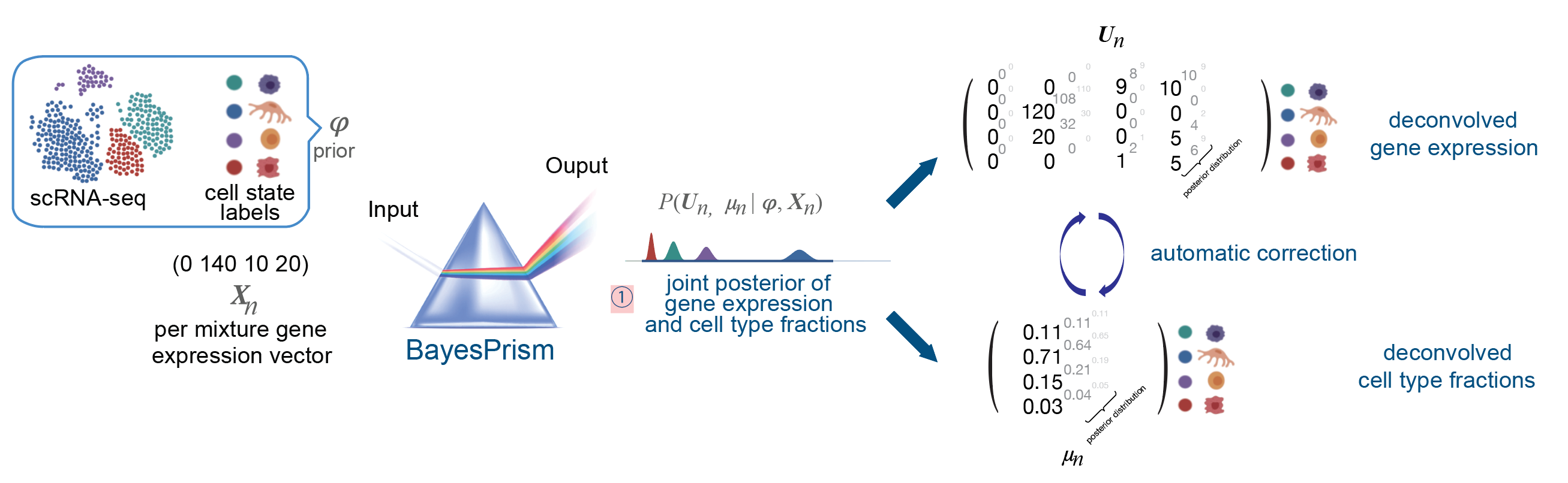
We develop statistically rigorous methods to disentangle cell-type composition and gene expression from bulk and spatial RNA-seq mixtures. Our flagship tool BayesPrism provides a fully Bayesian framework that jointly infers cell-type fractions and cell-type-specific expression — published in Nature Cancer 2022, where it has become the most-cited primary research paper in the journal since 2022 with 642+ citations, reflecting its broad adoption for tumor microenvironment analysis. Ongoing work extends BayesPrism into a next-generation deconvolution architecture that integrates eQTL modeling to characterize cell type-specific genetic effects and gene regulatory programs — including cis-eQTL dissection — bridging transcriptome deconvolution with population-level genetic analysis.
Nature Cancer 2022
Nature Genetics 2022
Ongoing
BayesPrism: scRNA-seq + bulk RNA-seq → joint posterior P(U,μ|φ,X) → deconvolved cell-type fractions & gene expression per sample.
🗺️
Spatial Transcriptomics and Cell–Cell Interaction Modeling
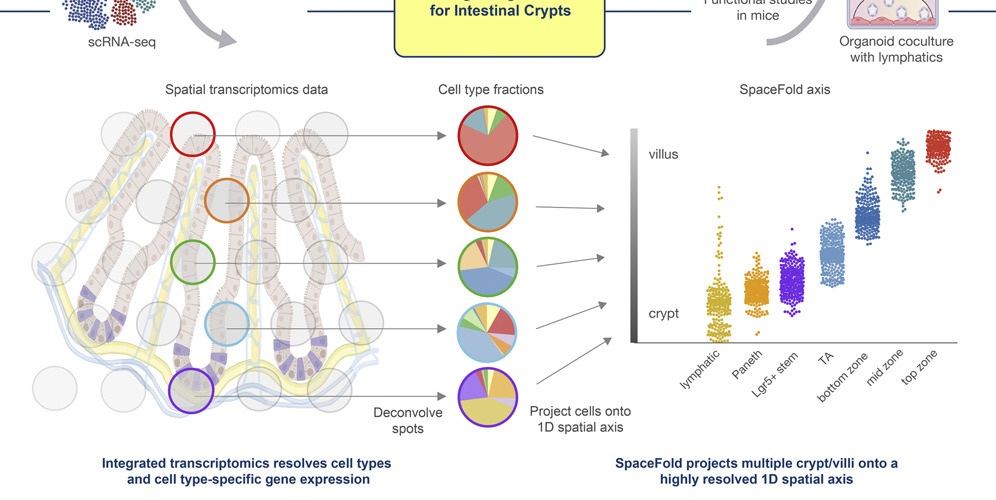
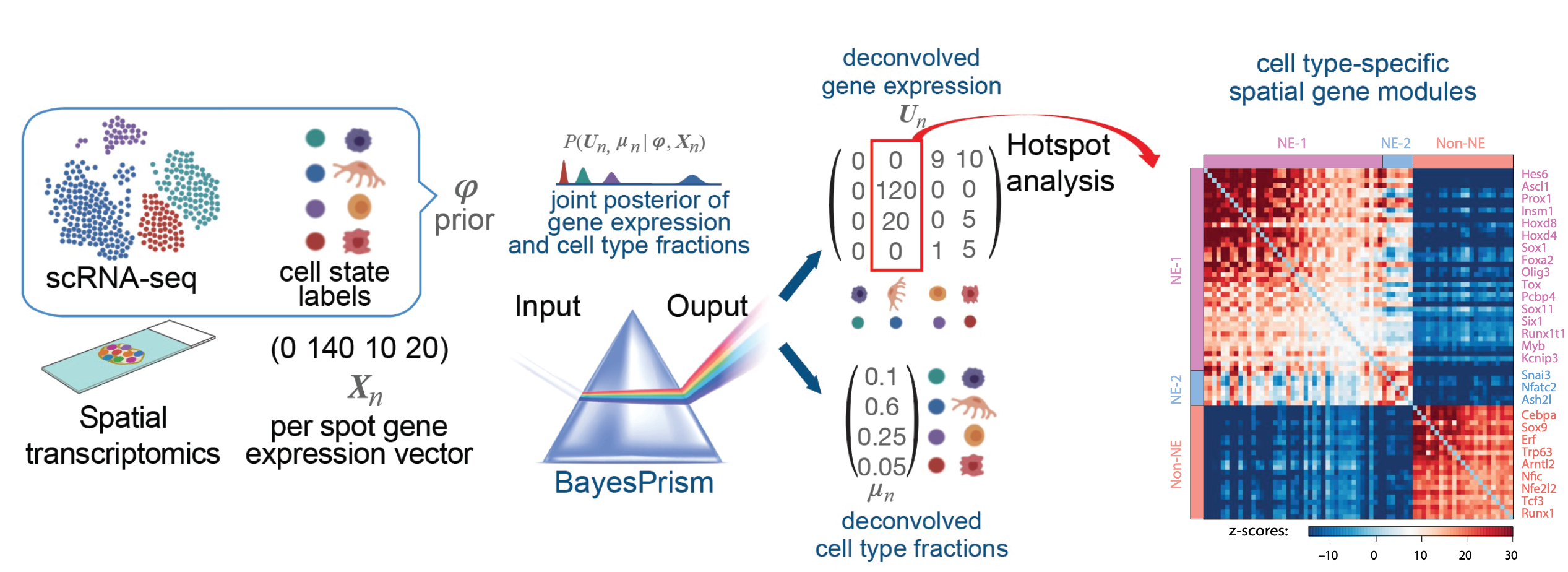
Tissues are not homogeneous — spatial context fundamentally shapes cell identity and behavior. We build methods that extract high-resolution gene expression cartography (SpaceFold), identify spatially variable gene programs within specific cell types (PrismSpot), and decode tissue microenvironments. Ongoing work develops deep learning frameworks to model cell–cell interactions directly from spatial transcriptomics data, capturing ligand–receptor signaling, paracrine communication, and niche-dependent gene regulation across complex tissues.
Cell Stem Cell 2022
Nature Cancer 2024
Science Immunology 2022
Ongoing
Top: SpaceFold maps 30+ cell types along the crypt–villus axis (Cell Stem Cell 2022). Bottom: PrismSpot identifies spatially variable gene programs within specific cell types (Nature Cancer 2024).
💧
Cell-free RNA & Liquid Biopsy
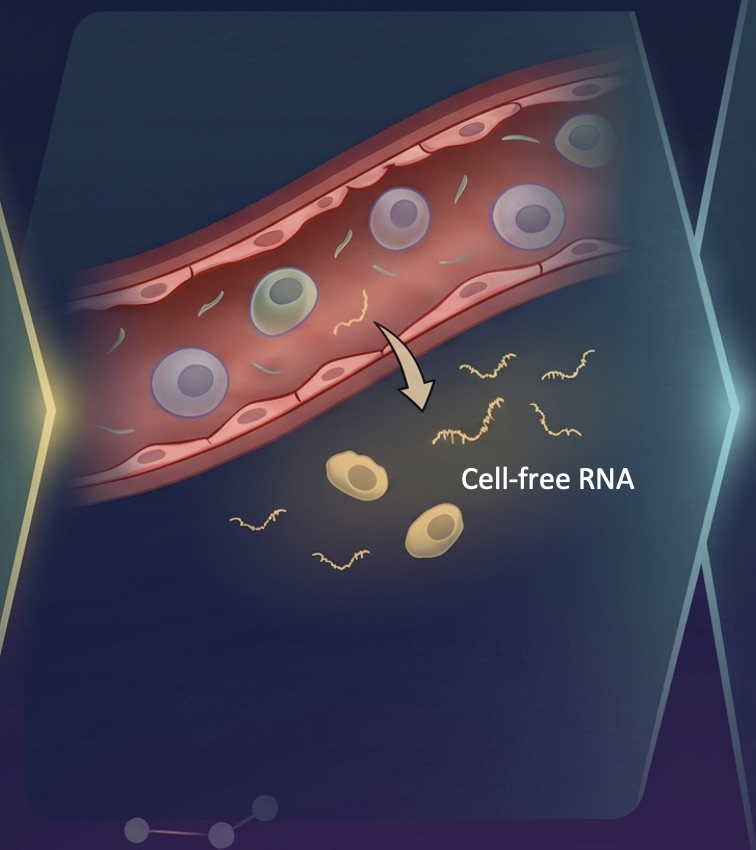
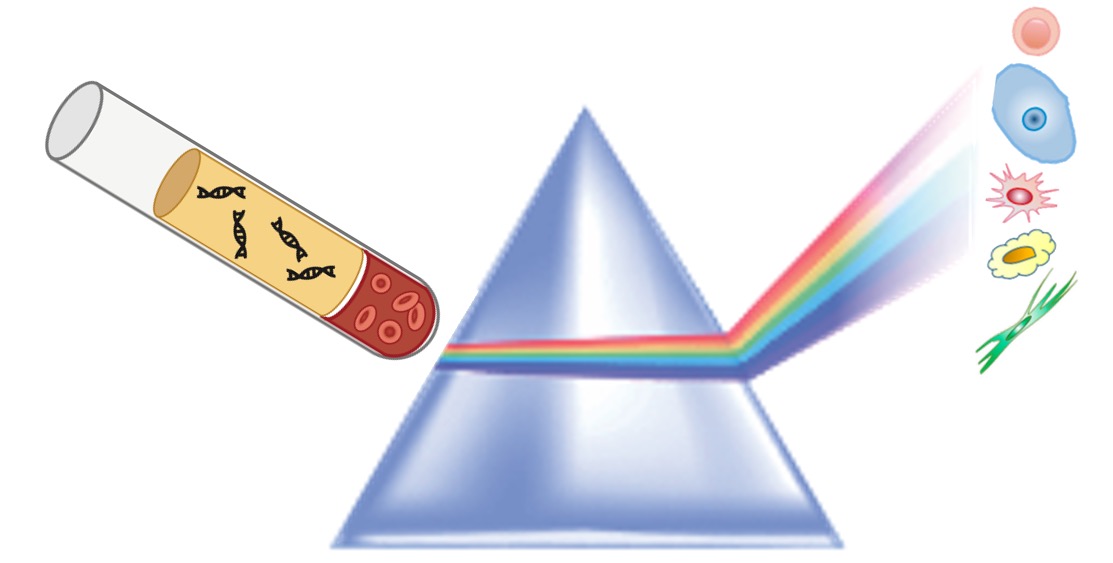
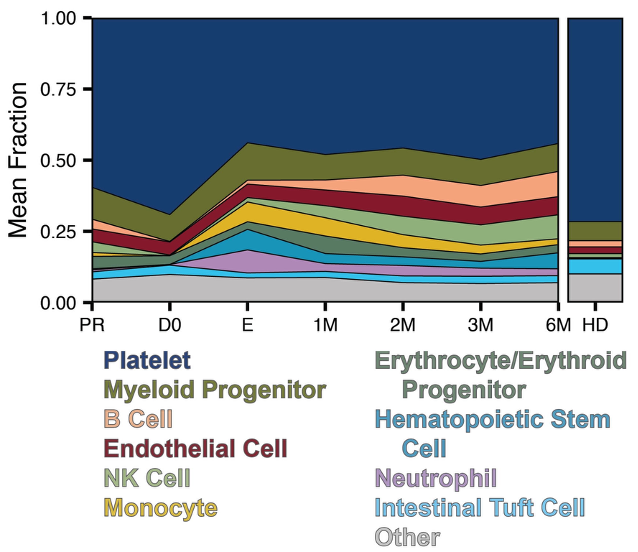
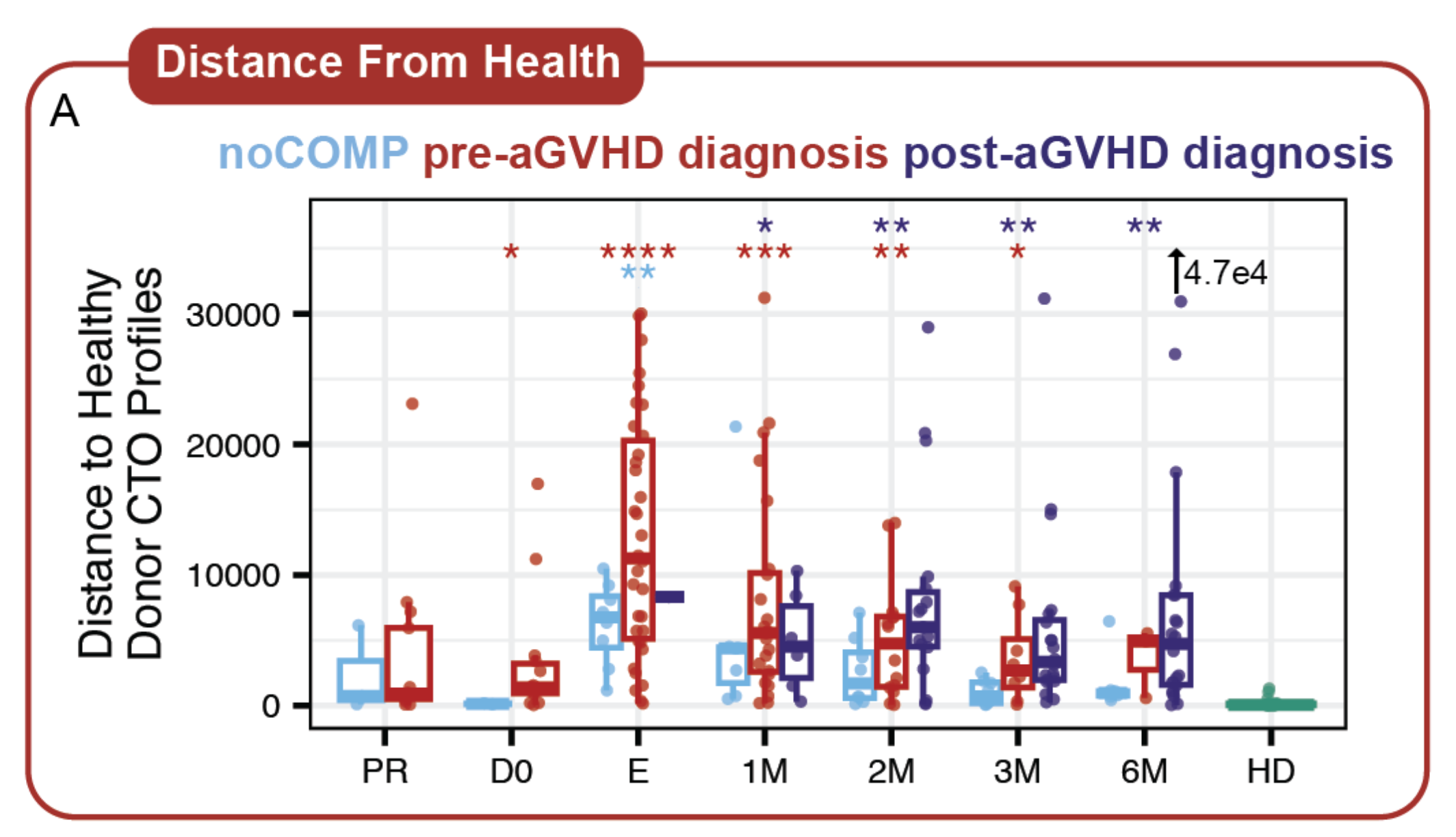
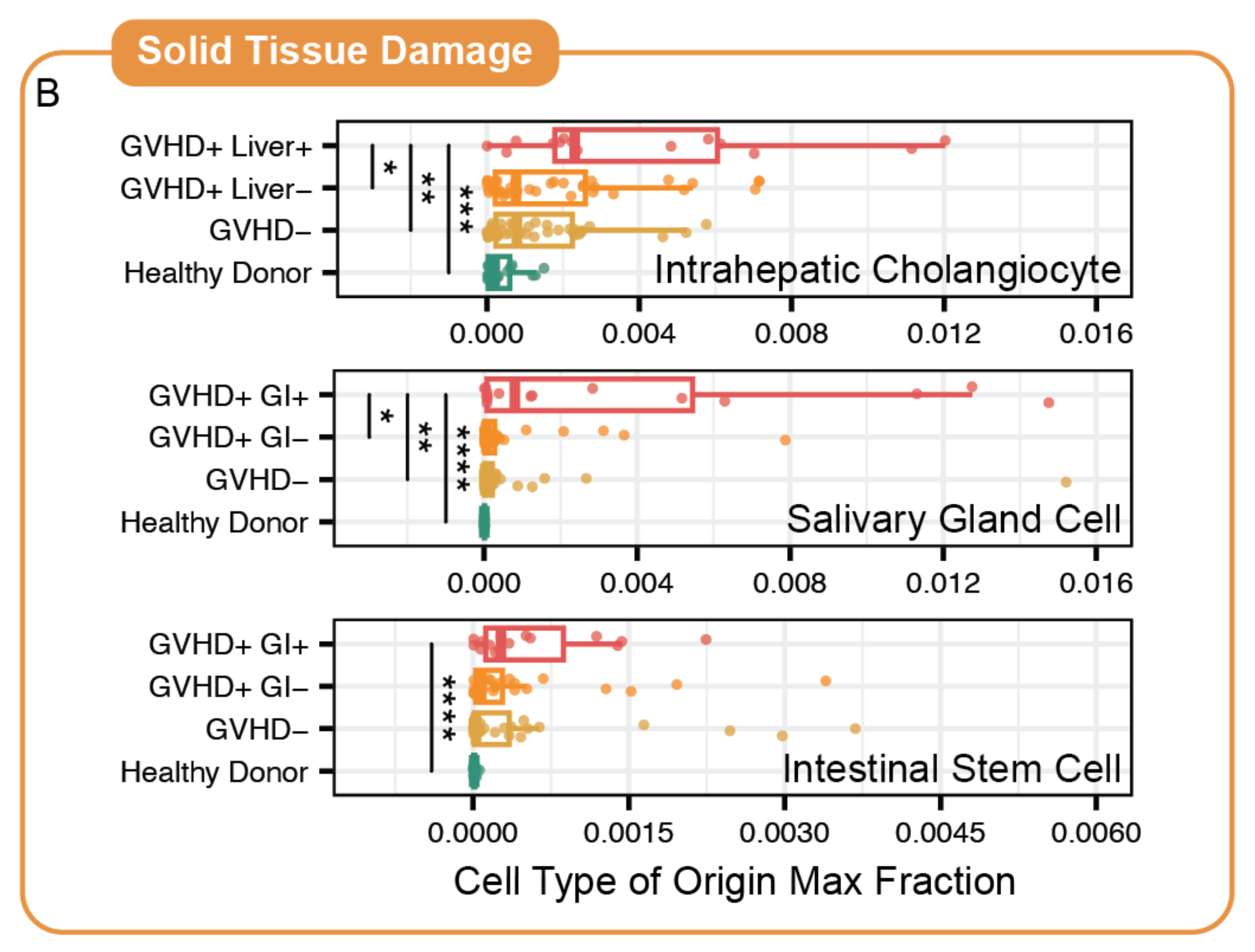
Cell-free RNA circulating in blood and urine carries molecular signatures of organ health. We develop statistical methods to infer cell-type origin from cfRNA, enabling non-invasive monitoring of organ-specific damage — demonstrated in hematopoietic stem cell transplantation and immune complications. This work carries significant translational potential: a simple blood or urine draw could replace invasive biopsies for monitoring transplant rejection, early cancer detection, and inflammatory organ injury across multiple diseases. Ongoing work expands into multimodal integration of cfRNA, cfDNA, and clinical variables, and develops methods to overcome the unique sparsity and distributional challenges inherent to liquid biopsy data.
medRxiv 2024
Under Review 2024
Top: cells shed RNA into circulation — cfRNA carries multi-organ cell-type signatures. Grid: cell-type fractions shift dynamically across transplant phases; distance-from-health predicts complications; cell-type origin pinpoints damaged organs.
〜
Modeling the Dynamics of Cell State Transitions
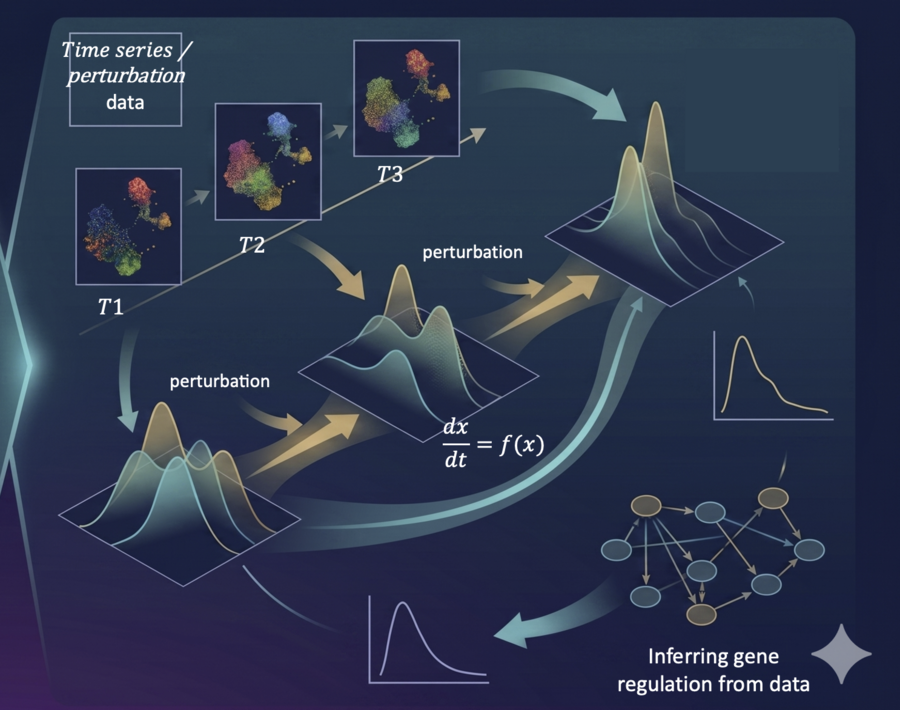
Gene expression is a dynamic process, yet most methods only analyze steady-state snapshots. We are developing neural differential equation and continuous-time dynamical models that learn the governing dynamics of gene regulatory networks from perturbation data — CRISPRi/a screens, cytokine treatments, spatial gradients, and time-series single-cell experiments. The ultimate goal is to identify causal regulators of cell state transitions: moving beyond correlation to pinpoint the master transcription factors and signaling nodes that drive or block specific cell fates, enabling rational design of in silico perturbations and virtual cell experiments.
NeurIPS / ICML Scope
Perturbation Genomics
Time-series / perturbation single-cell data (T1→T2→T3) → Neural ODE dx/dt=f(x) → infer gene regulatory dynamics and predict perturbation responses.